Module 3 | Lesson 5 | Fly-CURE - Converting, Sorting, and Indexing bam files
Overview
Teaching: 14 min
Exercises: 240+ minQuestions
How do I convert sam to bam files to sort and index the alignment?
Objectives
Understand why we convert from sam to bam.
Describe the steps involved in using samtools on our bam files.
Recorded Lesson:
Module 3 | Lesson 5 | Fly-CURE - Converting, Sorting, and Indexing bam files
Copying data to local directory
If this is a continuing analysis and you still have your FlyCURE/ directory, skip to SAM/BAM format.
If this is a new analysis, complete the following commands before proceeding. These are the only directories needed for the next lesson.
$ cd
$ GOCMD_VER=$(curl -L -s https://raw.githubusercontent.com/cyverse/gocommands/main/VERSION.txt); \
curl -L -s https://github.com/cyverse/gocommands/releases/download/${GOCMD_VER}/gocmd-${GOCMD_VER}-linux-amd64.tar.gz | tar zxvf -
The second step is to connect GoCommands to your account.
$ ./gocmd init (hit enter)
iRODS Host [data.cyverse.org]: (hit enter)
iRODS Port [1247]: (hit enter)
iRODS Zone [iplant]: (hit enter)
iRODS Username: (enter your CyVerse username)
iRODS Password: (enter your CyVerse password and hit enter. *You will not see the text as you are typing.)
The last step is to copy the data you need from the datastore to a local directory. To minimize the data transfer, each lesson will contain the path to grab only the data or directory you need. Although it seems like more work, it will take less time to upload using this method.
$ mkdir -p FlyCURE/results/bwa_out
$ mkdir -p FlyCURE/scripts
$ ./gocmd sync i:/iplant/home/your_username/FlyCURE/results/bwa_out ./FlyCURE/results
All paths in this lesson assume you are working in the local FlyCURE/ directory.
SAM/BAM format
The SAM file, is a tab-delimited text file that contains information for each individual read and its alignment to the genome. While we do not have time to go into detail about the features of the SAM format, the paper by Heng Li et al. provides a lot more detail on the specification.
The compressed binary version of SAM is called a BAM file. We use this version to reduce size and to allow for indexing, which enables efficient random access of the data contained within the file.
We are going to be utilizing a series of tools from the program Samtools which will eventually allow us to identify SNPs unique to each mutant. Samtools allows for manipulating alignment files in the BAM format. The imported file to Samtools is the SAM (Sequence Alignment/MAP) format which will be sorted, merged, and indexed allowing us to more easily retrieve reads in our region of interest. Link to manual pages for Samtools.
There are a series of 7 for loops that we will run. I will describe each step first.
Step 1: SAM to BAM Conversion.
Samtools only works with the BAM file format, and thus we must first convert our SAM to BAM using samtools view. We will be utilzing for loops to minimize errors and improve efficiency. By defining variables early in the process, we can use these same variable names in all of our for loops rather than utilizing individual sample names (Figure 1). In addition to the working components of the for loop, we will also add echo commands which will just provide us with a visual cue that our commands are running.
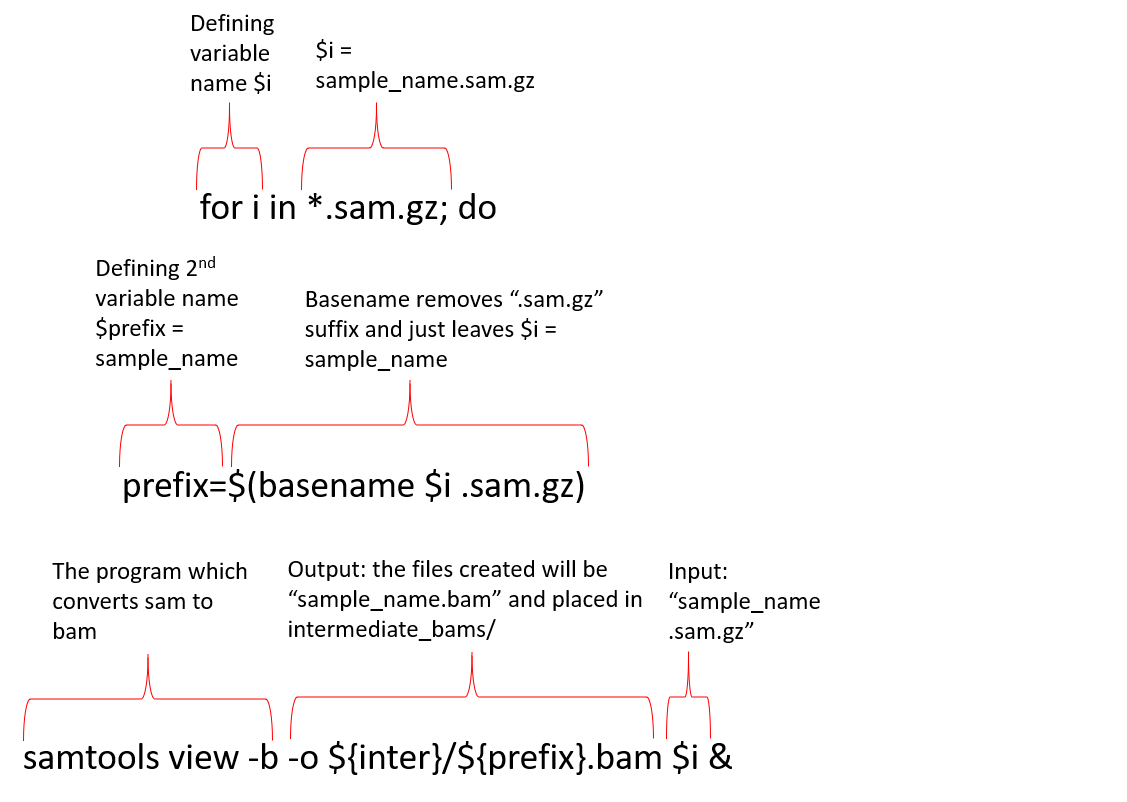 Figure 1: Breakdown of a
Figure 1: Breakdown of a for loop. This example provides a breakdown of the samtools view for loop. The formula for the for loop will remain mostly the same as we proceed through each of the samtools steps.
Step 2: Sort BAM by name
We will use samtools sort twice during our analysis. For this sort, we are going sort our BAMs by name. This will generate our reads in alphabetical order with the read name at the top of the file. Name sorting is required for the next step in the process.
Step 3: Fixmate on BAMs
Next we will use samtools fixmate which only works on name sorted BAMs. Fixmate will fix any errors that were made while aligning the paired-end reads.
Step 4: Sort BAM by coordinates
Once the paired-end mates are fixed, we will use samtools sort a second time this time sorting by genomic coordinates. This is the default sorting method of samtools sort.
Step 5: Mark and remove PCR duplicates
PCR duplicates are removed by a random sampling of the genome using samtools markdup. If you do random sampling of a genome, it is not likely you would see a bunch of sequences starting and ending at the same locations due to how the libraries for sequencing are generated. If there are samples that are represented at a statistically higher rate, these samples were likely overamplified, thus we remove them from the analysis to reduce our bias.
Step 6: Index the markdup BAMs
Once we have removed the marked duplicates from our data set, we then index the alignments using samtools index. Indexing allows for a more efficient way to search for sequences of interest just as an index of a book helps you find the topic of interest more quickly.
Step 7:
Lastly, we will calculate statistics of our reads using samtools flagstat.
To build the script, we are going to link the seven for loops together by using the wait command between each loop. When wait is used, the next loop will not begin until the sample has finished the first loop and so on. We also need to define some directories that we want our outputs sent to and where our inputs are located. I have started the script with commenting out the steps as a reminder to ourselves. The next set of comments describe where we want to launch the script, how we can log and watch the progress of the script, and the names we are going to use for our 2 output directories. We will want our results to land in FlyCURE/results. Our bwa_out directory is located in this location. Thus, we are going to create one directory called intermediate_bams to store results in the middle of the process and a second directory called clean where our final results will be output to. We have to give the relative path from where we are located when we launch the script. We start here: FlyCURE/results/bwa_out thus, to achieve this FlyCURE/results/intermediate_bams and FlyCURE/results/clean we can give the path of ../intermediate_bams and ../clean. It’s standard notation to indent the lines between the for and the done to give you a visualization of what is in the for loop.
Create the script by opening nano and copying and pasting the script from here into your text file.
$ cd FlyCURE/scripts
$ nano bam_factory.sh
#!/bin/bash
# what I do:
# step 1: sam to bam conversion
# step 2: sort bam by name (samtools sort -n)
# step 3: fixmate on bam
# step 4: samtools sort by coordinate
# step 5: mark duplicates +remove (samtools markdup -r)
# step 6: indexes the markdup bam
# step 7: calculates the stats on that file
# run me in the folder with sam.gz (FlyCURE/results/bwa_out)
# if you want to save literal commands to a log file, run me like
# ../../scripts/bam_factory.sh > mylogfile.txt
# therefore,
# intermediate_bams folder is defined below as $inter
# clean folder is defined below as $clean
inter='../intermediate_bams'
mkdir -p $inter
clean='../clean_bams'
mkdir -p $clean
#this loop converts sam to bam
for i in *.sam.gz; do
echo "converting $i to bam file"
prefix=$(basename $i .sam.gz)
echo samtools view -b -o ${inter}/${prefix}.bam $i
samtools view -b -o ${inter}/${prefix}.bam $i &
done
wait
# sorting by name loop
for i in *.sam.gz; do
echo "name sorting $i"
prefix=$(basename $i .sam.gz)
echo samtools sort -n -o ${inter}/${prefix}.nsort.bam ${inter}/${prefix}.bam &
samtools sort -n -o ${inter}/${prefix}.nsort.bam ${inter}/${prefix}.bam &
done
wait
# fix mate loop
for i in *.sam.gz; do
echo "fixmate $i"
prefix=$(basename $i .sam.gz)
echo samtools fixmate -r -m ${inter}/${prefix}.nsort.bam ${inter}/${prefix}.fixmate.bam &
samtools fixmate -r -m ${inter}/${prefix}.nsort.bam ${inter}/${prefix}.fixmate.bam &
done
wait
# re-sort by coordinate
for i in *.sam.gz; do
echo "coordinate sorting $i"
prefix=$(basename $i .sam.gz)
echo samtools sort -o ${inter}/${prefix}.csort.bam ${inter}/${prefix}.fixmate.bam &
samtools sort -o ${inter}/${prefix}.csort.bam ${inter}/${prefix}.fixmate.bam &
done
wait
# markdup & remove pcr duplicates loop
for i in *.sam.gz; do
echo "markdup removing dupes $i"
prefix=$(basename $i .sam.gz)
echo samtools markdup -r ${inter}/${prefix}.csort.bam ${clean}/${prefix}.markdup.bam &
samtools markdup -r ${inter}/${prefix}.csort.bam ${clean}/${prefix}.markdup.bam &
done
wait
# index loop
for i in *.sam.gz; do
echo "indexing $i"
prefix=$(basename $i .sam.gz)
echo samtools index ${clean}/${prefix}.markdup.bam &
samtools index ${clean}/${prefix}.markdup.bam &
done
wait
# what I do:
# run flagstat on an indexed bam file and write that stat report to a log
# sorting by name loop
for i in *.sam.gz; do
echo $index $bam to $log
prefix=$(basename $i .sam.gz)
echo samtools flagstat ${clean}/${prefix}.markdup.bam \> ${clean}/${prefix}.markdup.flags.log &
samtools flagstat ${clean}/${prefix}.markdup.bam > ${clean}/${prefix}.markdup.flags.log &
done
Make the script executable, navigate to bwa_out, and run the script!
$ chmod +x bam_factory.sh
$ cd ~/FlyCURE/results/bwa_out
$ ../../scripts/bam_factory.sh > bam_factory.log &
The & at the ends of the commands just lets the script run in the background so we could continue to use the terminal if we wanted/needed to. That also means your terminal prompt will return even though the script is still running. This isn’t all that important in our JupyterLab environment because we can open another terminal if needed. We can watch the progress of the script by using the command below or opening a second terminal and using top. Expect this script to take a few hours to run. If you want to exit tail or the log press q. (This should work, but we did find that you may have to shut down the terminal to get out of tailing the log.)
$ tail -F bam_factory.log
Completed file sizes when checked with ls -lh.
total 43G
-rw-r--r-- 1 gea_user gea_user 2.1G Feb 21 21:33 A44.markdup.bam
-rw-r--r-- 1 gea_user gea_user 548K Feb 21 21:41 A44.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 A44.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.7G Feb 21 21:37 B-1-3.markdup.bam
-rw-r--r-- 1 gea_user gea_user 549K Feb 21 21:42 B-1-3.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 B-1-3.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.9G Feb 21 21:38 B-2-13.markdup.bam
-rw-r--r-- 1 gea_user gea_user 549K Feb 21 21:42 B-2-13.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 B-2-13.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.7G Feb 21 21:37 B-2-16.markdup.bam
-rw-r--r-- 1 gea_user gea_user 551K Feb 21 21:42 B-2-16.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 B-2-16.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.7G Feb 21 21:37 Control.markdup.bam
-rw-r--r-- 1 gea_user gea_user 551K Feb 21 21:42 Control.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 Control.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 3.5G Feb 21 21:39 F-1-4.markdup.bam
-rw-r--r-- 1 gea_user gea_user 551K Feb 21 21:42 F-1-4.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:45 F-1-4.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.3G Feb 21 21:35 E-2-2.markdup.bam
-rw-r--r-- 1 gea_user gea_user 548K Feb 21 21:42 E-2-2.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 E-2-2.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.6G Feb 21 21:37 G-3-4.markdup.bam
-rw-r--r-- 1 gea_user gea_user 550K Feb 21 21:42 G-3-4.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 G-3-4.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 3.9G Feb 21 21:40 H22.markdup.bam
-rw-r--r-- 1 gea_user gea_user 553K Feb 21 21:42 H22.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:45 H22.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.5G Feb 21 21:37 H-3-2.markdup.bam
-rw-r--r-- 1 gea_user gea_user 549K Feb 21 21:42 H-3-2.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 H-3-2.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 3.7G Feb 21 21:39 L31.markdup.bam
-rw-r--r-- 1 gea_user gea_user 552K Feb 21 21:42 L31.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:45 L31.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 3.0G Feb 21 21:38 L-3-2.markdup.bam
-rw-r--r-- 1 gea_user gea_user 549K Feb 21 21:42 L-3-2.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:45 L-3-2.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.9G Feb 21 21:38 N-1-1.markdup.bam
-rw-r--r-- 1 gea_user gea_user 550K Feb 21 21:42 N-1-1.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 N-1-1.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 3.0G Feb 21 21:38 N-1-4.markdup.bam
-rw-r--r-- 1 gea_user gea_user 550K Feb 21 21:42 N-1-4.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:45 N-1-4.markdup.flags.log
-rw-r--r-- 1 gea_user gea_user 2.5G Feb 21 21:37 O-2-2.markdup.bam
-rw-r--r-- 1 gea_user gea_user 550K Feb 21 21:42 O-2-2.markdup.bam.bai
-rw-r--r-- 1 gea_user gea_user 443 Feb 21 21:44 O-2-2.markdup.flags.log
Exercise
Using the example I provided above for the samtools view
for loop, breakdown one (or more) of the otherfor loopssuch as for samtools sort or samtools fixmate. You can define it such as I do and/or replace the variables with a sample name to help you track what they all stand for.Solution
Figure 2: Breakdown of a
for loopfor samtools sort.If you were to write the
for loopfor a single sample it would look like this (don’t run the script as you already have):for i in A44.sam.gz do samtools sort -n -o intermediate_bams/A44.nsort.bam intermediate_bams/A44.bam doneNotice that I did not need to use
basenamebecause I was using the sample name itself. Alternatively, the for loop doesn’t even need to be used in this case and you could just use the samtools sort command directly.
Save analysis back to the data datastore
Since we were working locally, you will want to save your work to the datastore for future access. DO NOT FORGET THIS STEP.
$ cd
$ ./gocmd sync --single_threaded FlyCURE/results/clean_bams i:/iplant/home/your_username/FlyCURE/results
Note: The --single_threaded flag reduces the number of TCP connections (aka communication between devices over a network) to 1 thread which will create 4 TCP connections. The assumption is the amount of TCP connections we are creating is overwhelming our environment. By reducing the number of threads and TCP connections this “should” prevent the syncs from failing. This is also dependent upon how many people are trying to do this at the same time. Fewer threads means the sync will be slower. If you want, you can also use the flag --thread_num 0 and replace the number with 1 or 2 or 3 or 4 to increase the number of threads and therefore the processing speed, but you increase failure by increasing threads. Each thread creates 4 TCP connections.
Key Points
Bioinformatic command line tools are collections of commands that can be used to carry out bioinformatic analyses.
To use most powerful bioinformatic tools, you’ll need to use the command line.
There are many different file formats for storing genomics data. It’s important to understand what type of information is contained in each file, and how it was derived.